
Correct answer: b.
Discussion
Most CRCs develop from a series of genetic and/or epigenetic alterations in the epithelium, leading to the formation of adenomas. Inactivation of the adenomatosis polyposis coli (Apc) gene is present in more than 80% of CRCs.2,3 The multiple intestinal neoplasia (ApcMin/+) mouse, which carries a truncation mutation at codon 850 of the Apc gene, was the first key model of CRC. Like most genetically engineered mouse models, the ApcMin/+ mouse model has been largely used in studies of CRC development and chemoprevention, as part of a translational approach. However, its use in preclinical drug discovery is very limited. In addition, the short lifespan of the ApcMin/+ mouse and the low metastatic rate make this model unsuitable for mimicking advanced CRC.4 Still, in recent years, many different Apc mutant mice with germline or inducible conditional alleles of Apc have been developed, such as the ApcCKO/CKO-LSL-Kras mice (G12D; Krastm4tyj/+ allele), which exhibit simultaneous inactivation of Apc and activation of Kras in the adult colon, leading to the formation of distant metastases.3,5 These genetically modified Apc mouse models allow for more accurate preclinical investigations, including screening for early disease biomarkers.
The establishment of 3D culture systems from intestinal stem cells for the growth and maintenance of tissues explanted from intestinal and colonic mouse and human samples is thought of by many as the next big thing in CRC research.6 The method for creating and maintaining self-renewing intestinal organoids was pioneered by Sato and co-workers in 20097 and has since been optimized.8,9 There are several useful applications of organoid systems in CRC research:6
- Performing stem cell assays—functional evaluation of the stem cell capacity of distinct cell populations.
- Easy genetic manipulation—sequential deletion of a series of genes, as a way to model the multistep CRC development process, is possible by using the clustered regularly interspaced short palindromic repeats (CRISPR)–CRISPR-associated 9 (Cas9) system. For instance, isogenic human organoids that model the adenoma–carcinoma sequence in human CRC can be generated by editing of Apc by CRISPR–Cas9, followed by deletion of SMAD4 and p53 and, further, by introduction of activating mutations in KRAS and PIK3CA.10,11,12 Furthermore, modelling the genetic alterations associated with CRC carcinogenesis also allows genetic events associated with CRC stem cell generation to be studied.
- Studying poorly explored interactions between different cell types—it is possible to co-culture organoids from intestinal or colorectal adenomas with different cell types, such as lymphocytes, nerves and fibroblasts.
- Comparing drug or chemotherapeutic responses between tumour spheroids and normal intestinal epithelial organoids, as a readout of drug efficacy. Furthermore, such testing can easily be scaled up to high-throughput screening (HTS) of drugs, while patient-derived organoids allow for personalized therapy assays, like testing for individual responses to both existing and potential new drugs. This type of testing is of particular value for patients carrying rare mutations. Last but not least, organoids can theoretically be cultured from different regions of the CRC tumour, to model tumour heterogeneity, and from primary and metastatic locations to study mechanisms of cancer metastasis.10
Patient derived xenografts (PDXs), in which portions of a patient’s tumour tissue are implanted into an immunodeficient mouse, preserve, at least temporarily, the patient’s specific tumour–stromal-cell interactions. As such, PDXs were seen as a promising way to test therapies ahead of patient treatment, to screen for efficacy and resistance. However, although PDXs may predict clinical response to therapy better than traditional xenografts, they also have several disadvantages. For a start, host cells appear to replace the human stroma and vasculature much more quickly than initially estimated. In addition, the model requires subculture or serial transplants, which is expensive, labour intensive and time consuming (requiring ≥6 months).3 Furthermore, similarly to traditional xenografts, PDXs still need to be implanted into immunodeficient host animals, while its tumour initiation rate has been suggested to be ~70%, further limiting clinical utility.10,13
A syngraft or isograft model involves the implantation of cell lines or tumour fragments derived from a particular species into an immunocompetent animal of the same strain. The biggest advantage of such a model is its ability to test immunotherapies. In fact, the role of the immune system in CRC development appears to be particularly important.14 As an example, Bindea and co-workers recently demonstrated that for CRC the CXCL13 chemokine positively correlates with disease-free survival; they then placed MC38 cells (mouse colon adenocarcinoma-derived cells) in mice lacking the CXCL13 receptor CXCR5. Strikingly, MC38 isografts were more proliferative and displayed higher growth rates in Cxcr5–/– mice than in wild-type animals, underscoring the prognostic value of CXCL13 in assessing CRC tumour burden.15 Furthermore, the rodents used in isograft models are cheaper and more robust compared with their immunodeficient counterparts. Still, it should be noted that tumours in this model will express the rodent homologues of human tumour genes, which may limit the testing of targeted therapies.4




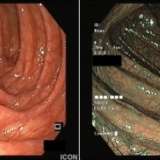
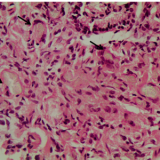
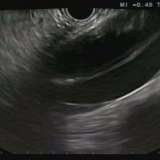
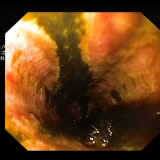
Please log in with your myUEG account to post comments.